Winkelwagen
U heeft geen artikelen in uw winkelwagen
Figure 1. Skin lesions in lymphomatoid papulosis. (A) Widespread erythematous papulonodular eruptions with scaling of the lesions (reproduced from Kavvalou et al. [45]). (B) Typical manifestation of lymphoid clumps (authors’ archive). (C) Typical manifestation with clustered nodules on the patient’s skin (authors’ archive).
Figure 1. Skin lesions in lymphomatoid papulosis. (A) Widespread erythematous papulonodular eruptions with scaling of the lesions (reproduced from Kavvalou et al. [45]). (B) Typical manifestation of lymphoid clumps (authors’ archive). (C) Typical manifestation with clustered nodules on the patient’s skin (authors’ archive).
PLEVA, one of pityriasis lichenoides variants, is an uncommon inflammatory skin disease, which may potentially be malignant. It generally presents with an acute-to-subacute eruption of multiple erythematous papules with hemorrhagic necrosis and crusting and often heals with atrophic varioliform scars. The lesions are frequently self-healing, however, recalcitrance may occur. 1 To confirm the diagnosis, histopathologic examination is required. PLEVA is characterized by interface dermatitis with prominent lymphocytic infiltration and epidermal involvement. 4 The exact pathogenesis remains unclear, however, immune dysregulation against medications/infectious agents or an evolution to cutaneous T-cell dyscrasia is an accepted hypothesis. 1 , 4 Despite lacking standard treatment, topical corticosteroids are commonly prescribed as the first-line therapy. Phototherapy, especially NB-UVB, and antibiotics including tetracyclines and erythromycin are also recommended. Low-dose methotrexate and other systemic immunosuppressants are indicated for severe or recalcitrant PLEVA. 5
The Clinicopathological Features and Immunohistochemistry Between Pityriasis Lichenoides Et Varioliformis Acuta, Lymphomatoid Papulosis, and Mycosis Fungoides
Notes: a Immunohistochemistry results: + indicates positive staining, − indicates negative staining, ± indicates positive or negative staining.
Abbreviations: LyP, lymphomatoid papulosis, MF, mycosis fungoides, PLEVA, pityriasis lichenoides et varioliformis acuta.
We presented a unique case of recalcitrant PLEVA for one year, and developed LyP after 3 months of remission, demonstrating two diseases in one patient. The association between PLEVA and LyP has been proposed but remains debatable. 11 , 18 However, clinicopathological and immunohistochemical evidence in the past decade has suggested PLEVA and LyP as distinct disorders. 4 , 19 , 20 Currently, there are limited reports of PLEVA and LyP arising in the same patient. 21 , 22 Sidiropoulou et al reported the coexistence of LyP type A, PLEVA, and MF over a 15-year period in one case. Different alterations in host immunity leading to discrete clinical expressions were remarked for these three separate entities. 21 Another case presented with prolonged LyP type B for 11 years followed by PLEVA, reflecting the difference in host immune response to antigenic stimulus. 22
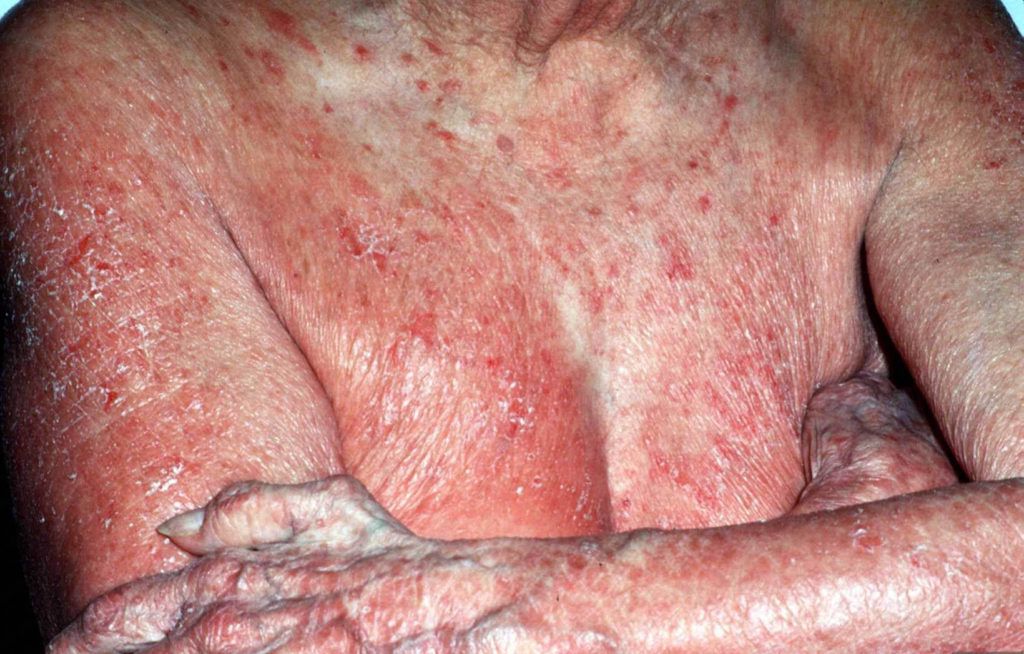
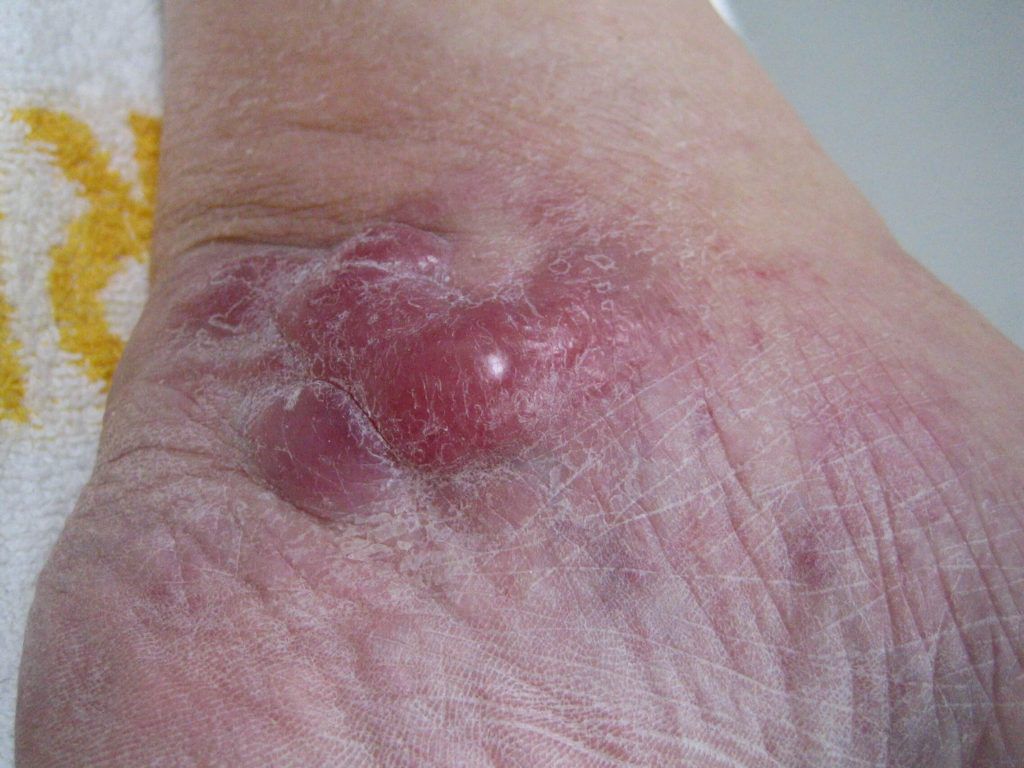
Lymphomatoid papulosis (LyP) is a very rare disease that belongs to the group of CD30+ lymphoproliferative skin diseases. LyP is localized or generalized and usually presents as isolated or clustered red/brown-red lesions in the form of nodules and/or papules. The course of the disease is in most cases mild, however, depending on concomitant risk factors and history, it may progress to lymphoma, significantly reducing the survival rate and prognosis. Importantly, the clinical picture of the disease remains somewhat ambiguous, leading to a large number of misdiagnoses that result in inappropriate treatment, which is usually insufficient to alleviate symptoms. In addition to clinical manifestations, the histological characteristics vary widely and usually overlap with other conditions, especially those belonging to the group of lymphoproliferative disorders. Although diagnosis remains a challenge, several recommendations and guidelines have been introduced to standardize and facilitate the diagnostic process. This article reviews the available literature on the most important aspects of etiopathogenesis, clinical and histopathological features, diagnostic criteria, and possible treatment strategies for LyP, with particular emphasis on the role of the immune system.
Lymphomatoid papulosis (LyP) is a relatively rare disease, occurring in the range of 1.2 to 1.9 cases per million people per year [1]. The data in the literature show that it is characterized by a bimodal peak incidence, the highest of which occurs in the 4th and 5th decade of life, especially in women, while the second, smaller peak occurs in children up to 18 years of age, with a higher percentage of male patients [2]. LyP presents with lesions that look like small red or red-brown bumps only a few millimeters in diameter, or they may appear as spots on the skin. As the disease progresses, the lesions may evolve into larger nodules and/or plaques and/or papules, usually with a maximum diameter of 2 cm, or they may go into spontaneous remission [3]. The etiology and pathogenesis of LyP remains unclear and is the subject of many interdisciplinary studies, including the mechanisms of spontaneous remission of the disease. Scientists are looking for evidence of involvement in the etiopathogenesis of onocogenic viruses (EBV, herpes virus), the participation of atopy (observed in about 50% of patients), genetic factors and susceptibility (aneuploidy and chromosomal aberrations), and abnormalities in the immune system [4,5].
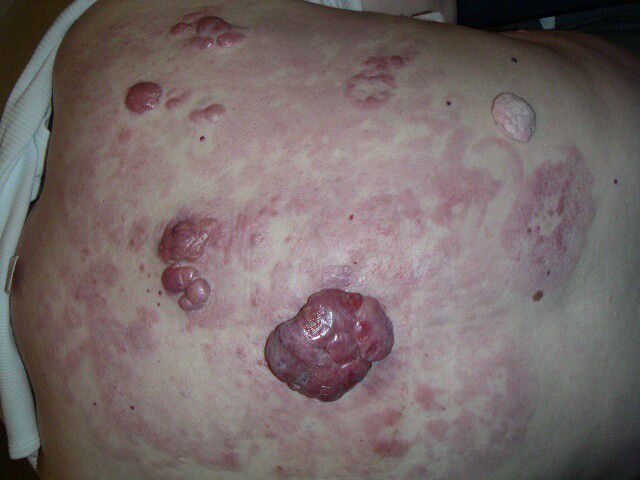
Currently, LyP may represent up to 12% of all diagnosed skin lymphomas [19,20]. It is the most frequently diagnosed skin lymphoma in the fourth and fifth decade of life in all ethnic groups equally [21,22]. The most important risk factor for LyP is HD or CTCL or a medical history of these two conditions, while the most common lymphomas associated with LyP are MF (24–61.4%), primary cutaneous ALCL (13–44%), and HD [21,23,24,25,26]. The risk of developing LyP-associated lymphoma is 2 to 7.5 times higher in patients with the occurrence of a monoclonal rearrangement of the TCR-γ (T-cell receptor) gene chain in skin lesions [26,27]. LyP has a diversified clinical and histological picture, while the misdiagnosis rate is difficult to determine, it is estimated to be about 30%, frequently resulting in unnecessary antibiotic treatment, chemotherapy, or radiotherapy [21,25]. LyP type A is the most diagnosed, with an estimated incidence of 47.2–82% compared to the other subtypes [12,21,26,28]. It has a 5-year survival of 100% [5,26,29], however, the prognosis may be reduced by the increased risk for progression to lymphomas, which is estimated to be 10–20% for LyP with adult-onset and 10% in the pediatric population [23,26].
Molecular and histological aspects must be discussed together with clinical presentation. The genetic instability of tumor cells is responsible for several characteristics. First, patients must be monitored for the development of lymphoma. Second, a patient may present with more than one histological subtype of LyP. Finally, the disease can progress or resolve spontaneously. All patients with LyP require long-term control visits, twice a year, to evaluate their clinical presentation, as described below.
LyP is a chronic disease of cutaneous lymphoid infiltration, characterized by a diverse clinical morphology and the occurrence of skin lesions such as papules, plaques, and nodules [30]. Early lesions appear as small red or reddish-brown or red-violet nodules a few millimeters in diameter that may be singular, clustered, or generalized [31]. As they grow, these lesions can develop into larger nodules and plaques, usually with a maximum diameter of no more than 1–2 cm [32,33,34]. Although complete regression may occur within a few weeks, papules may also progress to sterile pustules or become necrotic and then lead to hemorrhagic scabs and varioliform atrophic scarring [35,36]. In patients with intermittent LyP ejection, changes may coexist in different stages of development, resulting in a differentiated and polymorphic clinical picture [15]. Skin lesions can occur anywhere on the body, however, they usually develop mainly on the limbs and torso and, less often, on the face [37]. There are only a few reports of oral or genital involvement in the literature [38,39].
Histopathologic features of primary cutaneous anaplastic large cell lymphoma. (A) Sheets of large lymphoma cells with extensive dermal and subcutaneous tissue involvement with no epidermotropism. (B) Angiocentric distribution may be seen in some cases and mimic other angiotropic lymphomas (see text). (C) Sheets of large atypical lymphoid cells with prominent nucleoli replacing the dermis. An eccrine duct (center) is seen surrounded by tumor cells.
Histopathologic features of primary cutaneous anaplastic large cell lymphoma. (A) The papillary dermis is replaced by sheets of large pleomorphic atypical cells with abundant amphophilic to basophilic cytoplasm and kidney-shaped nuclei (“hallmark” cells). (B) Higher magnification of a “hallmark” cell. (C) The lymphoma cells may occasionally contain abundant cytoplasmic vacuoles. (D) Pseudoepitheliomatous hyperplasia in a case of primary cutaneous anaplastic large cell lymphoma.
Histologic variants of primary cutaneous anaplastic large cell lymphoma. (A) The “neutrophil/eosinophil-rich variant” of anaplastic large cell lymphoma, also known as pyogenic cutaneous lymphoma, may mimic a reactive inflammatory or infectious process (see text). (B) The “small cell variant” is extremely challenging to initially recognize as an anaplastic large cell lymphoma. (C) Primary cutaneous anaplastic large cell lymphoma with numerous apoptotic bodies can resemble extranodal NK/T-cell lymphoma or cutaneous gamma-delta T-cell lymphoma. (D) “Lymphohistiocytic variant” of anaplastic large cell lymphoma may be confused with a histiocyte-rich inflammatory process or an infection by an atypical mycobacteria.