Winkelwagen
U heeft geen artikelen in uw winkelwagen
An idiosyncratic, delayed hypersensitivity reaction has been implicated in the pathophysiology of Stevens-Johnson syndrome. Certain population groups appear more susceptible to develop Stevens-Johnson syndrome than the general population. Slow acetylators, patients who are immunocompromised (especially those infected with HIV [3, 4] ), and patients with brain tumors undergoing radiotherapy with concomitant antiepileptics are among those at most risk.
Antigen presentation and production of tumor necrosis factor (TNF)–alpha by the local tissue dendrocytes results in the recruitment and augmentation of T-lymphocyte proliferation and enhances the cytotoxicity of the other immune effector cells. [7] A "killer effector molecule" has been identified that may play a role in the activation of cytotoxic lymphocytes. [8] The activated CD8+ lymphocytes, in turn, can induce epidermal cell apoptosis via several mechanisms, which include the release of granzyme B and perforin.
In 1997, Inachi et al demonstrated perforin-mediated apoptosis in patients with Stevens-Johnson syndrome. [9] Perforin, a pore-making monomeric granule released from natural killer cells and cytotoxic T lymphocytes, kills target cells by forming polymers and tubular structures not unlike the membrane attack complex of the complement system.
Apoptosis of keratinocytes also can take place as a result of ligation of their surface death receptors with the appropriate molecules. Those can trigger the activation of the caspase system, leading to DNA disorganization and cell death. [10]
Apoptosis of keratinocytes can be mediated via direct interaction between the cell-death receptor Fas and its ligand. Both can be present on the surfaces of the keratinocytes. Alternatively, activated T-cells can release soluble Fas ligand and interferon-gamma, which induces Fas expression by keratinocytes. [1] Researchers have found increased levels of soluble Fas ligand in the sera of patients with SJS/TEN before skin detachment or onset of mucosal lesions. [11]
Mittlerweile ist diese Begriffsverwendung allerdings obsolet, da für die beiden Hautreaktionen unterschiedliche Ätiologien bestehen. Stevens und Johnson gelten als Erstbeschreiber des Stevens-Johnson-Syndroms und haben dem Symptomkomplex seinen Namen gegeben.
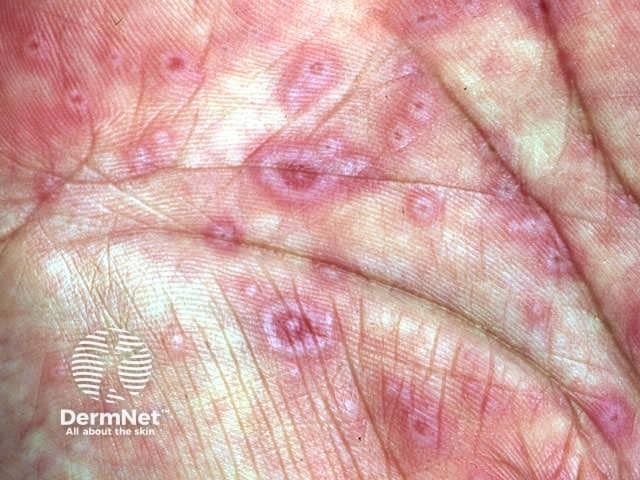
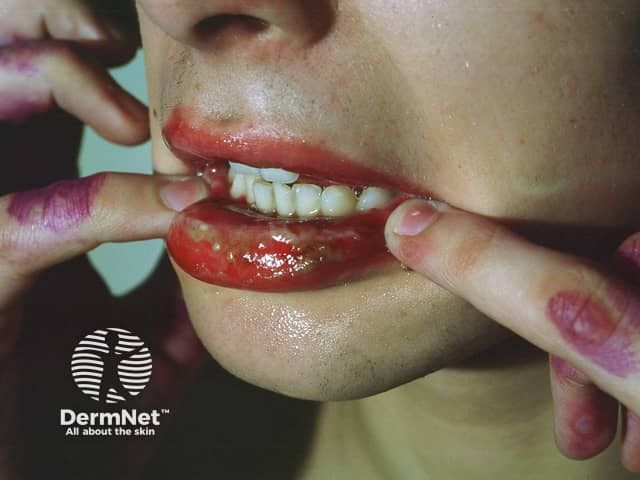
Das Stevens-Johnson-Syndrom kann sehr schwer verlaufen. Die Geschwüre lassen im Verlauf der Abheilung Narben zurück. Auch Schleimhautschrumpfungen können auftreten. Da die Schleimhäute in der akuten Krankheitsphase sehr gereizt sind, besteht das Risiko einer Zweitinfektion mit lokalen Erregern, wie zum Beispiel Pilze oder Bakterien.
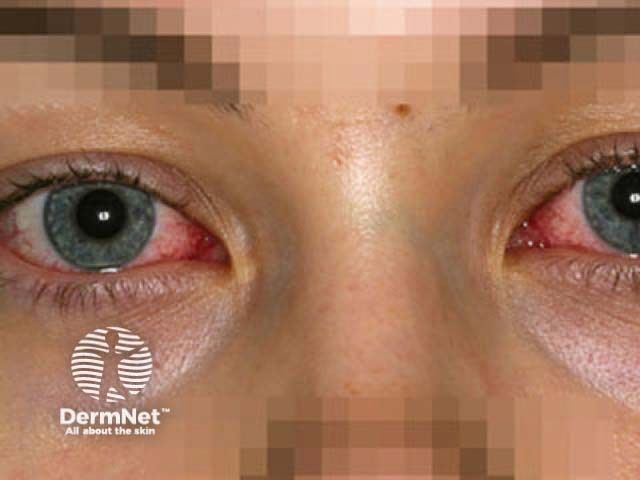
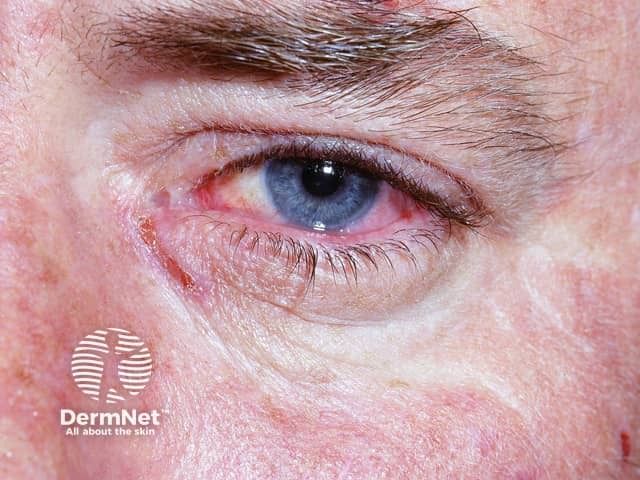
Des Weiteren kann es zu einem starken Flüssigkeitsverlust und in der Folge zu Dehydration und körperlichen oder geistigen Ausfallerscheinungen kommen. Greift die Entzündung auf die Augenhaut über, kann dies in einer Bindehautentzündung resultieren. Eine schwere Komplikation ist das Lyell-Syndrom, in dessen Verlauf sich die Haut ablöst und nekrotisch vernarbt.
In einem von vier Fällen endet das Folgesymptom tödlich. In weniger schweren Fällen bleiben Haut-Pigmentstörungen zurück. Weitere Komplikationen können durch Verwachsungen und Veränderungen in der Schleimhautstruktur auftreten. Die Therapie des Stevens-Johnson-Syndrom birgt verschiedene Risiken – etwa durch Präparate wie Makrolid-Antibiotika und Tetracycline.
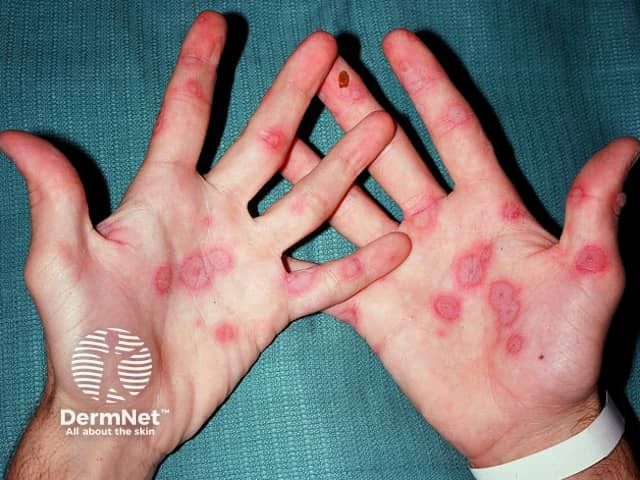
The syndrome was first described in 1922, when the American pediatricians Albert Mason Stevens and Frank Chambliss Johnson reported the cases of 2 boys aged 7 and 8 years with "an extraordinary, generalized eruption with continued fever, inflamed buccal mucosa, and severe purulent conjunctivitis." Both cases had been misdiagnosed by primary care physicians as hemorrhagic measles.
Erythema multiforme (EM), originally described by Ferdinand von Hebra in 1866, was part of the differential diagnosis in both cases but was excluded because of the "character of skin lesions, the lack of subjective symptoms, the prolonged high fever, and the terminal heavy crusting." Despite the presence of leukopenia in both cases, Stevens and Johnson in their initial report suspected an infectious disease of unknown etiology as the cause.
In 1950, Thomas divided EM into 2 categories: erythema multiforme minor (more common and as previously described by von Hebra) and erythema multiforme major (EMM). Since 1983, erythema multiforme major and Stevens-Johnson syndrome had been considered synonymous.
In the 1990s, however, Bastuji and Roujeau each proposed that erythema multiforme major and Stevens-Johnson syndrome are 2 distinct disorders. [2] They suggested that the denomination of erythema multiforme should be restricted to patients with typical targets or raised edematous papules, with or without mucosal involvement. This clinical picture is in accordance with the original description by von Hebra.
Bastuji and Roujeau further proposed that the denomination of Stevens-Johnson syndrome should be used for a syndrome characterized by mucous membrane erosions and widespread small blisters that arise on erythematous or purpuric maculae that are different from classic targets.
According to this clinical classification, erythema multiforme major and Stevens-Johnson syndrome could be 2 distinct disorders with similar mucosal erosions, but different patterns of cutaneous lesions. This hypothesis is supported further by a strong correlation between clinical classification and the probable cause.